
信达PD-1闯关FDA失败,14:1不建议批准上市
2月10日晚间,信达/礼来PD-1抗体信迪利单抗正式迎来了FDA肿瘤药物专家委员会(ODAC)的审评会议,探讨基于中国临床数据的信迪利单抗治疗非小细胞肺癌的联合疗法能否在美国获批上市。值得注意的是,这也是国产PD-1肿瘤药首次直面ODAC,闯关FDA,被业界认为是中国创新药凭借中国临床试验数据出海的“试金石”。
最终,会议以14:1的投票结果要求信达生物补充额外的临床试验,即信达/礼来PD-1不被建议直接获批。华尔街见闻报道称,虽然ODAC评议的结果不是FDA最终是否批准的全部依据,但FDA历史上基本会遵从专家委员会的评议结果,这也意味着信迪利单抗在美国的上市申请暂时遇挫。
争议点
FDA提出异议的主要原因包括以下几点:
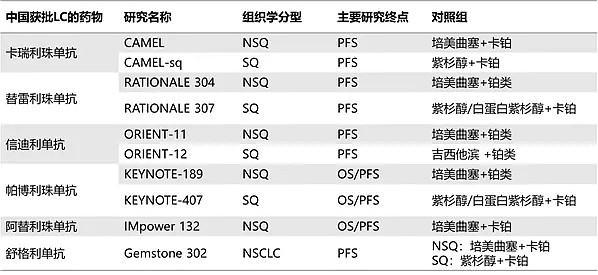
1、ORIENT-11不是一个国际多中心临床研究,与ICH E17指导原则描述不相符。
信迪利单抗所基于的ORIENT-11三期临床是一项于2018年8月开始的仅在中国进行而非国际多中心临床研究(MRCT)的试验,旨在评估信迪利单抗注射液或安慰剂联合培美曲塞和铂类用于晚期或复发性非鳞状非小细胞肺癌(NSCLC)一线治疗有效性和安全性的随机、双盲、3期对照临床研究(ClinicalTrials.gov, NCT03607539)。
FDA认为ORIENT-11不符合ICH E17指导原则的描述。ICH E17泛指多区域临床试验计划与设计的一般原则,从有着20年历史的E5(种族因素)演化而来,于2017年生效,以提高MRCT在全球监管递交中的可接受度。
对此,礼来申辩认为,信迪利单抗的临床试验在中国48个具有肿瘤学专业知识、大量患者的学术中心开展,且FDA曾进行过17次检查:12次检查没有发现任何问题,4次观察问题通过设计纠正得到解决,1次结果待定。这意味着ORIENT-11临床研究的质量符合要求。
FDA针对此举出中国CDE 2016年发布的新药申请数据造假等历史问题,怀疑ORIENT-11可能会有影响。提问环节,FDA明确在已调查的临床试验中,未发现任何欺诈或数据不完整问题;但其强调:正因为检查范围有限,才更要求多区域临床试验。
2、不符合美国医疗实践的对比组和终点(PFS)
ORIENT-11以无进展生存期(PFS)为临床终点,FDA表示OS(总生存期)是肿瘤临床试验中最可靠的疗效终点,应当以OS为临床终点。
在国内,根据CDE于2019年发布的《晚期非小细胞肺癌临床试验终点技术指导原则》,随着治疗手段的丰富,OS不断延长增加了评价难度,因此单独PFS或PFS与OS共同终点可被接受作为初治晚期NSCLC注册研究的主要终点。中国现在所有已批的PD-1单抗一线肺癌的终点都是PFS。
而近年来美国批准的NSCLC药物基本都以OS终点为主。
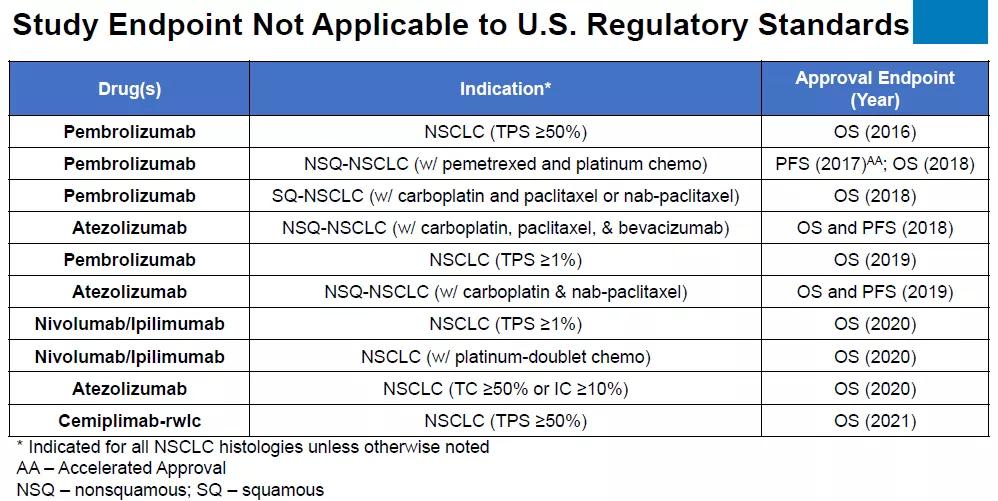
在此前的内部审查文件中,FDA曾批评ORIENT-11“在没有FDA咨询或监督的情况下,使用不符合美国监管标准或不符合美国医疗实践的对比组和终点(PFS)”而开展。
同时,FDA认为应该把已批准的PD-1疗法作为对照组进行头对头试验,这样才能确信这种新药物的疗效。2018年8月20日Keytruda联合化疗即获批用于治疗非鳞状NSCLC,NSCLC临床一线疗法已经从化疗变为免疫治疗+化疗联合治疗,而ORIENT-11选择的对照组为一线化疗方案。
此外,其他的不赞成点还包括:研究人群与美国人群存在已知和未知的内源性和外源性因素差异;ORIENT-11 缺乏与FDA的咨询和监督;根据GCP要求,知情同意书没有及时更新标准治疗选择;ORIENT-11 的适应症不是医疗迫切需求,不被授权监管灵活性等。
结果:14比1
在经历了近5个小时的观点陈述、问题澄清、公开讨论答辩等过程后,最终在关于"是否需要额外的适用于美国病人和美国医疗临床试验证明?"的投票环节,15位专家中,有14位专家选择“yes”,1位选择“no”。
选择“no”的南加州大学的Jorge Nieva教授,他表示“现有的临床数据证据足够支持有效性和安全性。对临床研究医师、研究机构和知情同意书的质疑,都没有直接证据。这些质疑可以是合理的,如果能证明与药品的有效性和安全性相关的话”。 使用交叉设计的PFS终点并没有硬伤,FDA检查并未找到实际作假证据,应该鼓励便宜、有效的治疗方法应该被鼓励进入美国。
此前,礼来曾在一个电话会议中表示出对于该PD-1有一个“颠覆性定价策略”,希望以价格优势让美国患者获益。据悉与市场上其他PD-1抑制剂相比,该药物的定价计划有40%的折扣。但此次FDA表示:“不会在监管决策中考虑药品定价,成本和药品定价不应作为ODAC会议的讨论主题”。
信达的回应
今日(2月11日),信达生物发布自愿公告,回应美国食品药品监督管理局(FDA)对信迪利单抗关于治疗一线非鳞非小细胞肺癌的新药申请召开肿瘤药物咨询委员会一事。信达生物在公告中表示,ORIENT-11是一项高质量、高标准、由经验丰富的临床研究者参与的符合全球认证GCP要求的中国临床试验。ORIENT-11试验结果数据展示了信迪利单抗的良好的风险获益关系。公司和礼来制药将继续配合FDA完成新药上市申请的审评工作。

以下为公告内容:
美国食物药品监督管理局(「FDA」)召开肿瘤药物咨询委员会(「ODAC」),对信迪利单抗注射液的新药上市申请(「BLA」)审评问题进行讨论并投票。此次BLA申报适应症为信迪利单抗注射液联合培美曲塞和铂类用于非鳞状非小细胞肺癌(「nsqNSCLC」)一线治疗,主要基于在中国开展的ORIENT-11临床三期试验资料。委员会投票建议需要在获批前补充额外临床试验,证明信迪利单抗在美国人群和美国医疗实践中的适用性。信迪利单抗是一款创新PD-1抑制剂,由本公司与礼来制药公司(「礼来」)共同开发和商业化。
ORIENT-11是一项高质量、高标准、由经验丰富的临床研究者参与的符合全球认证GCP要求的中国临床试验。ORIENT-11试验结果数据展示了信迪利单抗的良好的风险获益关系。FDA没有任何对于信迪利单抗安全性和有效性问题的质疑。
本公司和礼来制药将继续与FDA配合完成新药上市申请的审评工作。ODAC就已上市和临床中的肿瘤药品,为FDA提供独立的专业性意见。FDA在新药审批过程中将采纳ODAC的投票意见,但ODAC投票意见不具有对FDA决策的约束力。
本公司对信迪利单抗的临床和商业化价值一如既往充满信心。此外,此次申报与美国监管机构的深入沟通与交流,为本公司大大锻炼了海外注册团队,为创新管线的全球开发提供了大量的宝贵经验。公司将更加坚定加速布局管线的全球化发展和加大创新人才的全球布局,加速从生物科技公司(biotech)向全球生物制药公司(global biopharma)的转型,带动公司持续发展,将高品质的创新药物带向全世界的患者。



